

10 Jul XlinkX for Proteome Discoverer
Crosslinking mass spectrometry made easy
Recently, structural proteomics, and more specifically crosslinking mass spectrometry (XL-MS), gained a large amount of traction as a supplemental method to protein structure techniques like electron microscopy (EM), nuclear magnetic resonance (NMR) and crystallography. XL-MS generally provides distance constraints of lower resolution than these other techniques, but is able to pinpoint residues in close proximity to the interaction interfaces between individual subunits for protein complexes of any size in solution. Provided there are sufficient crosslinks, this technique can even allow for the detection and definition of protein interfaces. The information obtained from XL-MS experiments have in many cases been successfully leveraged to produce final protein complex models.

XlinkX for Proteome Discoverer (developed with support of the Proteome Discoverer development team) makes the analysis of XL-MS data easy with a user-friendly interface, highly advanced FDR control, visualization options for identified spectra and many other plots to investigate the results in detail. The software supports any linker (cleavable or non-cleavable, which can be fully defined in the general modification editor of Proteome Discoverer) and can handle any modifications (also definable in the general modification editor of Proteome Discoverer).
Below we supply workflows that are standard in use in our laboratory for XL-MS data analysis. For more information on acquiring xlinkx for PD, check out the Thermo Fisher Scientific webpage.
Features
XlinkX in Proteome Discoverer supports to following features:
- Any non-cleavable crosslinker (definable in the PD modifications editor)
- Any cleavable crosslinker (definable in the PD modifications editor)
- Crosslink on and between multiple possible amino acids (e.g. for NHS ester lysine to tyrosine)
- Search with any fixed or variable modification (maximum of 5; definable in the PD modifications editor)
- Search can be fully configures with regards to protease (definable in the PD protease editor), number of missed cleavages, minimum peptide length, maximum number of modifications allowed on one of the peptides, minimum and maximum peptide mass, precursor and fragment tolerances (Orbitrap and Ion Trap spectra are automatically treated with the required tolerance), and score filtering options.
- Blistering fast non-cleavable crosslinker search for databases up to 1500 proteins driven by sequence tags and immonium ions
- Search of limitless databases for cleavable linkers driven by diagnostic ion detection
- Advanced, machine learning driven FDR control of the results
- Efficient grouping of crosslinked peptide pair identifications for an aggregate view on the results
- Calculates crosslink occupancies
- Calculates PTM scores for the detected crosslink positions
Standard workflows
Proteome Discoverer 2.3
Additional tooling around XL-MS and structural biology in general
[coming soon] CrosslinkLab
This separate tool allows you to dive into the mass spectral data behind the identifications and is able to load either raw-data directly (a convenient interface is provided to define the peptide identities) or the Proteome Discoverer output directly. The spectra can be visualized and importantly exported to PDF format to support submission to journals like MCP.
Features:
- Spectral visualization
- Define any peptide pair with a user friendly editor (including all PTMs defined in Unimod)
- Score calculations of the spectral match
- Exports for PDF representation of the annotated spectra, annotated tab separated mass-lists, digest report of each fragment series, and many others
- Easy and fast search options to locate peptide-pairs / PTMs / spectra of interest in hundreds of thousands entries
- Option to define specific mass tolerances, signal to noise ratios and many others
[coming soon] CrossI’d
This separate tool allows you to visualize the complex datasets in network format and enables filtering, clustering and drilling down into the spectral data for fast and accurate interrogation of the results.
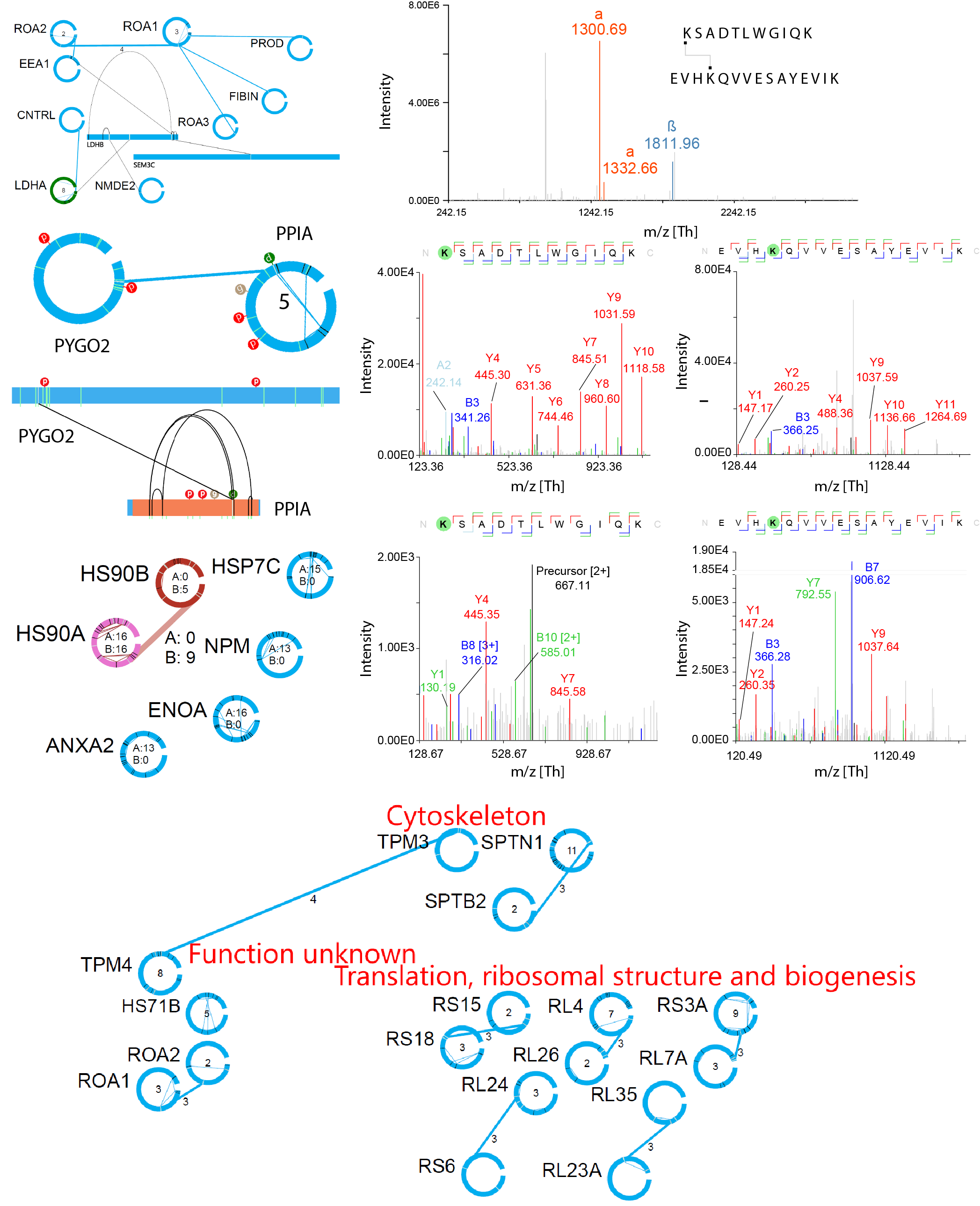
Features:
- Visualization of large networks of proteins connected with XL-MS
- Clustering based on GO annotations
- Incorporates the latest Uniprot information on the proteins
- Fast search option for specific proteins
- Allows to immediately drill down to the annotated spectral data to visually verify identifications
- Easy compare multiple datasets
- Visualization of quantitation data
- And many more
Download
[wpfilebase tag=file id=139 tpl=download-button /]
Cross-ID video
References
Klykov et al; Nature Protocols – in press; Efficient and robust proteome-wide approaches for crosslinking mass spectrometry
Liu et al; MCP 2018; The interactome of intact mitochondria by cross-linking mass spectrometry provides evidence for coexisting respiratory supercomplexes
https://www.ncbi.nlm.nih.gov/pubmed/29222160
Fagerlund et al; PNAS 2017; Spacer capture and integration by a type I-F Cas1-Cas2-3 CRISPR adaptation complex
https://www.ncbi.nlm.nih.gov/pubmed/28611213
Liu et al; Nat Communications 2017; Optimized fragmentation schemes and data analysis strategies for proteome-wide cross-link identification
https://www.ncbi.nlm.nih.gov/pubmed/28524877
Lössl et al; ACS Central Science 2016; Deciphering the Interplay among Multisite Phosphorylation, Interaction Dynamics, and Conformational Transitions in a Tripartite Protein System
https://www.ncbi.nlm.nih.gov/pubmed/27504491
Benda et al; Mol Cell 2014; Structural model of a CRISPR RNA-silencing complex reveals the RNA-target cleavage activity in Cmr4
https://www.ncbi.nlm.nih.gov/pubmed/25280103
![]()
Sorry, the comment form is closed at this time.